In contrast to other known cellulosomes, in B. cellulosolvens the cohesin types are reversed for all scaffoldins i.e., the type II cohesins are located on the enzyme-integrating primary scaffoldin, whereas the type I cohesins are located on the anchoring scaffoldins.
It has been previously shown that type I B. cellulosolvens interactions possess a dual-binding mode that adds flexibility to scaffoldin assembly. Here we show the structural mechanism of enzyme recruitment into B. cellulosolvens cellulosome and identify the molecular determinants of its type II cohesin-dockerin interaction mechanism.
The results suggest that, unlike other type II complexes, these possess a dual binding mode of interaction, similarly to type I.
Therefore, the plasticity of dual-binding mode interactions seems to play a pivotal role in the assembly of B. cellulosolvens
| "...in B. cellulosolvens the cohesin types are reversed for all scaffoldins ..."
The present study has revealed the structure (PDB code: 2y3n) and mechanism of the type II Coh-Doc complex interaction responsible for enzyme incorporation into the main scaffoldin ScaA of B. cellulosolvens cellulosome. The structure presented shows typical folds for both the cohesin and dockerin modules, similar to what is found in the vast majority of Coh-Doc structures.
Computational modelling as well as binding studies suggest that the two modules interact via a dual-binding mode, thus imparting the dual-binding mode as a general feature of all type I and II interactions in B. cellulosolvens. This means that the assembly of B. cellulosolvens’ cellulosome relies solely on cohesin-dockerin complexes with a dual-mode of binding.
Considering the incredible complexity of this system and the potential to assemble up to 110 enzymes in a single unit, it is not surprising that this cellulosome has evolved towards acquiring a high degree of flexibility and plasticity that would allow the accommodation of such a high number of both enzymatic and structural components in close proximity.
For more information:
Further reading:

During the course of evolution, the cellulosome, one of Nature’s most intricate multi-enzyme complexes, has been continuously fine-tuned to efficiently deconstruct recalcitrant carbohydrates.
To facilitate the uptake of released sugars, anaerobic bacteria use highly ordered protein-protein interactions to recruit these nanomachines to the cell surface. Dockerin modules located within a non-catalytic macromolecular scaffold, whose primary role is to assemble cellulosomal enzymatic subunits, bind cohesin modules of cell envelope proteins, thereby anchoring the cellulosome onto the bacterial cell.
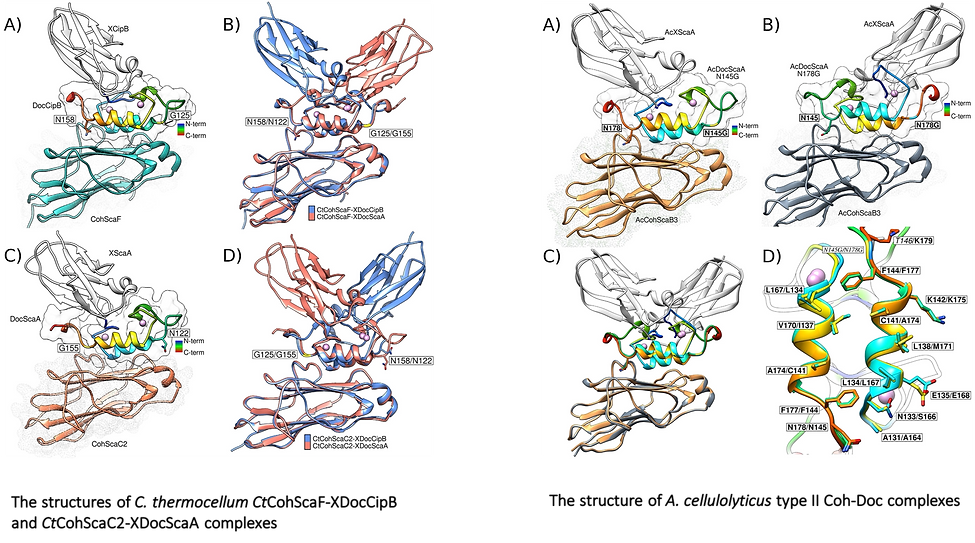
Here we elucidated the unique molecular mechanisms used by anaerobic bacteria for cellulosome cellular attachment. The structure and biochemical analysis of five cohesin-dockerin complexes revealed that cell surface dockerins contain two cohesin-binding interfaces, which can present different or identical specificities. In contrast to the current static model, we propose that dockerins utilize multivalent modes of cohesin recognition to recruit cellulosomes to the cell surface, a mechanism that maximises substrate access while facilitating complex assembly.
| "...cell surface dockerins contain two cohesin-binding interfaces, which can present different or identical specificities."
This study reveals that cellulolytic anaerobic bacteria, notably C. thermocellum and A. cellulolyticus, employ different molecular mechanisms to attach elaborate cellulosomal complexes to their bacterial cell surfaces. Type-II Docs of primary scaffoldins ScaA and CipB in C. thermocellum possess two distinct Coh-binding surfaces that recognise different Coh partners. These diverse specificities allow a single but distinct Doc binding mode to the different Cohs presented on the surface of this bacterium. In contrast, suppleness in Coh recognition is a dominant feature of the type-II Doc module of A. cellulolyticus, where a dual-binding mode was observed. The latter mechanism of Coh-Doc recognition would impose additional structural freedom to ensure efficient assembly of these elaborate complexes and the corresponding degradation of plant cell walls.
For more information:
Further reading:
Understanding the specific molecular interactions between proteins and β1,3-1,4-mixed-linked D-glucans is fundamental to harvest the full biological and biotechnological potential of these carbohydrates and of proteins that specifically recognise them. The family 11 carbohydrate-binding module from Clostridium thermocellum (CtCBM11) is known for its binding preference for β1,3-1,4-mixed-linked over β1,4-linked glucans. Despite the growing industrial interest of this protein for the biotransformation of lignocellulosic biomass, the molecular determinants of its ligand-specificity are not well defined.
The native structure of CtCBM11 was determined in 2004 to a resolution of 1.98 Å and is deposited in the PDB under the code: 1v0a [ref.]. Its structure suggested that the contacts between residues Ser59, Asp99, Tyr53, Arg126, Tyr129 and Tyr152 and the histidine tail of a symmetry-related molecule could impair ligand binding and thus co-crystallisation and soaking experiments. To tackle this problem I determined the crystal structure crystal structure of CtCBM11 without the histidine tag (structure not deposited). The new crystals belong to the P21 space group and comparison of the two structures reveals no major differences at the main-chain level and the two structurally relevant calcium atoms are conserved.
|"...comparison of the two structures reveals no major differences..."
Building up on previous data, we used an integrated approach combining carbohydrate microarrays, NMR, X-ray crystallography, site-directed mutagenesis and ITC to unravel, at a molecular level, the ligand recognition of CtCBM11. The analysis of the interaction by carbohydrate microarrays and NMR and the crystal structures of CtCBM11 bound to β1,3-1,4-linked glucose oligosaccharides, showed that both the chain length and the position of the β1,3-linkage are important for recognition, and identified the tetrasaccharide Glcβ1,4Glcβ1,4Glcβ1,3Glc sequence as a minimum epitope required for binding.
The bound structures of CtCBM11 were solved at a resolution of 1.45 Å and 2.6 Å for complexes with G4G4G3G (PDB 6R3M) and G4G3G4G4G3G (PDB 6R31). The structural data, along with site-directed mutagenesis and ITC studies, demonstrated the specificity of CtCBM11 for the twisted conformation of β1,3-1,4 mixed-linked glucans. This is mediated by a conformation-selection mechanism of the ligand in the binding cleft through CH-π stacking and a hydrogen bonding network, which is dependent not only on ligand chain length, but also on the presence of a β1,3-linkage at the reducing-end and at specific positions along the β1,4-linked glucan chain.
In the context of the cellulosome, the structural details revealed on the CtCBM11 ligand-recognition site may influence the planning and development of efficient and low-cost mechanisms for the conversion of biomass into usable sources of energy, as well as, into nutrients for animal feedstock.
Additionally, the understanding, at the molecular level, of how CtCBM11 selects and binds its ligands may inspire the design of new biomolecules with improved capabilities to be explored in health and agriculture applications.
For more information:
Further reading:

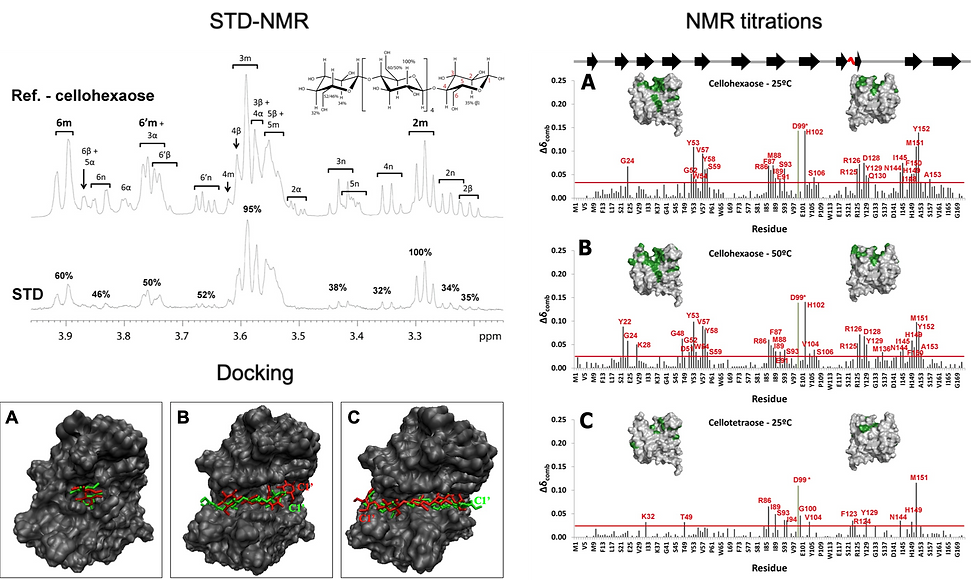
C. thermocellum grows at T_opt of 60 ºC and has T_max of 69 ºC and a Tmin above 28 ºC. In order to investigate the influence of temperature in the protein structure and dynamics we have determined the NMR solution structure of the protein at 25 and 50 ºC following a standard triple resonance approach using double labeled (13C and 15N) CtCBM11. The coordinates of the structures determined at 25 and 50 ºC were deposited in the BMRB data bank (http://www.bmrb.wisc.edu) (access numbers: 18388 and 18389, respectively) and in the PDB (http://www.pdb.org/pdb) (PDB codes: 2lro and 2lrp, respectively).
Both structures are very similar to each other, with an r.m.s.d. of 1.03 Å between the ensemble representative NMR solution structures. This is indicative of a very stable protein, as one would expect from a thermophilic organism. Also, both structures are similar to the X-ray structure, with r.m.s.d. of 1.20 Å for the structure at 25ºC and 1.10 Å, for the structure at 50ºC. However, a careful comparison between the NMR solution structures and the crystal structure shows that the β-sheet elements superpose quite well, whereas the loop regions superpose less well. This is especially true in the loop formed between residues R125-Q134, which has the largest r.m.s.d. value.
Interestingly, this makes that in the NMR structures the binding cleft area is larger than in the crystal structure (approximately 3700 and 3760 Å2 for the structures at 25 and 50 ºC versus 3225 Å2). The closed conformation of the binding cleft imposed by the crystal packing, as displayed in the X-ray structure, may impair the binding of cellooligosaccharides and the difference found between the solution and the X-ray structure might explain the failed attempts for co-crystallizing CtCBM11 with several ligands.
| "...in the NMR structures the binding cleft area is larger than in the crystal structure."
This result reveals a key role of the geometry of the binding cleft in the interaction with cellooligosaccharides. In this sense, NMR provides a more accurate description of the solution structure of CtCBM11 as it accounts for the conformational modifications of the binding cleft that allow ligand binding. The results indicate that significant changes in the binding cleft may occur do to the crystal packing and this is an important information to consider when using X-ray structures for binding studies, especially molecular docking studies.
For more information:

Molecular determinants of ligand specificity of the family 11 carbohydrate-binding module from Clostridium thermocellum (CtCBM11)
While the crystal structure of C. thermocellum CBM11 had been resolved in the apo form [ref.], no structure with a bound substrate could not be obtained at the time. Thus we used a combined approach by X-ray Crystallography, NMR and Computational Chemistry, in order to gain further insight into the binding mode of different carbohydrates (cellobiose, cellotetraose and cellohexaose) and understand the molecular recognition of glucose polymers by CtCBM11
These studies provided information on the residues of the protein involved in ligand recognition and on the influence of the length of the saccharide chain on binding. A cluster of aromatic residues has been found to be important for guiding and packing the polysaccharide. Linebroadening, STD-NMR and DOSY experiments allowed screening the binding activity of the several ligands and identifying the atoms of the ligands closer to the protein upon binding (epitope mapping). The binding cleft of CtCBM11 interacts more strongly with the central glucose-units of cellotetraose and cellohexaose, mainly through interactions with the OH groups at position 2 and 6 of the central sugar units.
The models of the CtCBM11/cellohexaose and CtCBM11/cellotetraose complexes obtained by docking allowed a detailed inspection of the main protein ligand interactions. CH-π and Van der Waals interactions were found to be determinant for the stability of the complexes and to the specificity of the protein. Protein relaxation data analysed in terms of the model-free approach revealed that the protein behaves as an axial symmetric rotor of the oblate type, independently of the state (bound or free) or temperature. Moreover, thermodynamic data extracted from the titration experiments at 25 and 50 ºC and from the general order parameter, S2, indicates that binding of cellooligosaccharides to CtCBM11 must occur by a “conformational selection” mechanism where the disposition of the residues in the binding cleft and interactions with specific groups of the ligand act as determinants of specificity in CtCBM11.
Altogether, the results presented allow an atomistic rationalisation of the molecular determinants of ligand specificity in CtCBM11 and the mechanism by which this protein is able to distinguish and select its ligands.
For more information:
Further reading: